Sindromul Joubert este o tulburare genetică rară în care creierul se dezvoltă anormal, ceea ce provoacă subdezvoltarea unei părți a creierului numită vermis cerebelos și malformații ale trunchiului cerebral; vermisul cerebelos coordonează mișcările capului, trunchiului și membrelor superioare, în timp ce trunchiul cerebral controlează funcții precum respirația, tensiunea arterială și ritmul cardiac.
În fiecare an, unul din 100.000 de copii din lume se naște cu sindrom Joubert. Majoritatea cazurilor sunt provocate de mutații genetice moștenite, dar afecțiunea se poate declanșa și din cauza unor mutatii genetice izolate/spontane.
Sindromul Joubert poate fi clasificat în mai multe subtipuri, în funcție de caracteristicile majore:
- clasic:
- probleme cu mișcarea ochilor;
- probleme respiratorii;
- degete suplimentare la mâini sau la picioare.
- cu boală retiniană:
- displazie de retină.
- cu boală renală:
- probleme cu rinichii.
- cu boală oculorenală:
- probleme de retină și de rinichi.
- cu boală hepatică:
- probleme hepatice.
- cu trăsături oral-facial-digitale:
- despicătură labio-palatină;
- probleme cu limba;
- degete suplimentare la mâini sau la picioare.
- cu caracteristici acrocalosale:
- apare în forme severe și presupune dezvoltarea deficitară a țesutului care leagă partea stângă și dreaptă a creierului.
- cu distrofie asfixiantă Juene:
- degete suplimentare la mâini sau la picioare;
- cutie toracică îngustă;
- statură mică.
Cauze
Sindromul Joubert se poate dezvolta din cauza mutațiilor genetice moștenite de la părinți sau a unor mutații genetice care se dezvoltă sporadic.
Au fost descoperite mutații în peste 40 de gene. Aceste mutații explică 94% din cazurile de sindrom Joubert, dar cauza genetică a celorlaltor șase procente rămâne încă necunoscută. Unele din cele mai frecvente cauze includ mutații în genele:
- AHI1;
- CEP290;
- NPHP1.
Mutațiile genetice care cauzează sindromul Joubert au ca rezultat anomalii ale cerebelului, trunchiului cerebral și cililor copilului:
- cerebelul gestionează coordonarea și mișcarea; în sindromul Joubert, vermisul cerebelos lipsește sau este mai mic decât în mod normal;
- trunchiul cerebral, care reglează respirația și echilibrul, nu se formează corect;
- cilii sunt structuri minuscule care ies în afara suprafețelor celulelor și le ajută să comunice între ele în timpul dezvoltării organelor.
Factori de risc
Sindromul Joubert are de obicei un model de moștenire autosomal recesiv, ceea ce înseamnă că, pentru a avea afecțiunea, o persoană trebuie să moștenească două gene modificate, câte una de la fiecare părinte.
O mutație rară a genei OFD1 asociată cu sindromul Joubert se găsește pe cromozomul X; femeile au doi cromozomi X, iar bărbații unul.
Femeile care suferă de această mutație a genei OFD1 au un risc de 25% să aibă:
- fiică ce poartă gena;
- fiică ce nu poartă gena;
- un fiu care are sindrom Joubert;
- un fiu care nu are sindromul Joubert.
Bărbații cu această genă au:
- 100% șanse de a transmite sindromul Joubert fiicelor lor;
- 0% șanse de a-l transmite fiilor lor.
Simptome
Semnele și simptomele sindromului Joubert sunt adesea evidente precoce în timpul copilăriei și pot varia foarte mult, dar includ în primul rând tulburări de coordonare, întârzieri de dezvoltare neuropsihomotorie și tonus muscular scăzut.
Semnele și simptomele afecțiunii mai includ:
- mișcare atipică a limbii sau a ochilor;
- apnee în somn;
- hiperpnee – respirație rapidă;
- incapacitatea de a coordona mișcările musculare;
- convulsii;
- anomalii ale rinichilor sau ficatului;
- meningoencefalocel (defect de sutură la nivelul craniului);
- probleme hormonale;
- anomalii ale limbii;
- probleme oculare.
Persoanele cu sindrom Joubert pot dezvolta o varietate de probleme de comportament legate de:
- controlul atenției;
- control emoțional;
- psihoză;
- aptitudini sociale.
Diagnosticare
Diagnosticul definitiv al sindromului Joubert este testarea genetică dar suspiciunea este ridicată de evaluarea clinică a pacienților care prezintă simptomele clasice de întârziere de dezvoltare neuropsihomotorie, tulburări de coordonare și hipotonie.
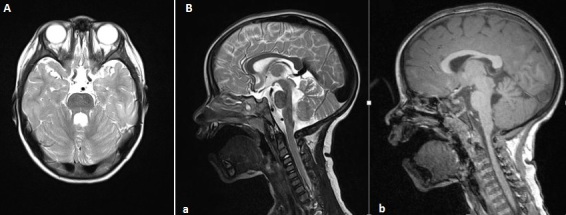
Din punct de vedere imagistic, poate fi indicată efectuarea unui RMN cerebral care evidențiază „semnul de dinte molar” pe RMN: testul imagistic va arăta că vermisul cerebelos este absent și că trunchiul cerebral este atipic dezvoltat, acest aspect semanand cu un dinte; aceasta se evidențiază printr-un model care seamănă cu un dinte.
Tratament
Tratamentul pentru sindromul Joubert este, de obicei, unul de susținere și are ca scop abordarea simptomelor specifice.
În managementul afecțiunii se recomandă o abordare multidisciplinară din partea unei echipe formate din neurologi, oftalmologi, nefrologi, geneticieni, kinetoterapeuți, logopezi, terapeuți ocupaționali și alții:
- un copil cu întârzieri intelectuale sau de dezvoltare poate urma terapie fizică, logopedie sau terapie ocupațională;
- dacă există probleme respiratorii, acestea pot necesita oxigen suplimentar, ventilație mecanică sau o traheostomie;
- în caz de anomalii oculare, pot fi necesare intervenții chirurgicale sau lentile corective;
- o disfuncție severă la nivel renal sau hepatic poate necesita dializă sau transplant.
Complicații
Sindromul Joubert poate dezvolta complicații pe mai multe paliere:
- la nivelul sistemului nervos central: unii copii pot avea convulsii, au fost raportate câteva cazuri de autism, iar tulburările de comportament pot include impulsivitate și crize de furie;
- la nivel scheletic: copiii cu un tonus anormal pot dezvolta complicații ortopedice, cum ar fi scolioza;
- la nivel oftalmologic: pot apărea strabism, ambliopie și boala retiniană;
- la nivel renal: se indentifică printre altele poliurie, anemie, insuficiență de creștere sau boala polichistică renală;
- la nivel hepatic: multe cazuri de fibroză hepatică ajung să necesite transplant.
Prevenție
Sindromul Joubert este o afecțiune genetică. În prezent, nu există o modalitate cunoscută de a o preveni, dar geneticienii și consilierii genetici pot ajuta la determinarea riscului la care sunt expuși membrii unei familii de a avea un copil cu această afecțiune.
Sursa: www.medlife.ro